Publications by EEOB 19-24 March 2014

Plethodon idahoensis, a salamander. Image: © Gary Nafis CaliforniaHerps.com | CC BY-NC-ND 3.0
Model Choice for Phylogeographic Inference using a Large Set of Models
Tara A. Pelletier and Bryan C. Carstens. 2014. Molecular Ecology in press. DOI: 10.1111/mec.12722
Abstract
Model-based analyses are common in phylogeographic inference because they parameterize processes such as population division, gene flow and expansion that are of interest to biologists. Approximate Bayesian Computation is a model-based approach that can be customized to any empirical system and used to calculate the relative posterior probability of several models, provided that suitable models can be identified for comparison. The question of how to identify suitable models is explored using data from Plethodon idahoensis, a salamander that inhabits the North American inland northwest temperate rainforest. First, we conduct an ABC analysis using five models suggested by previous research, calculate the relative posterior probabilities, and find that a simple model of population isolation has the best fit to the data (PP = 0.70). In contrast to this subjective choice of models to include in the analysis, we also specify models in a more objective manner by simulating prior distributions for 143 models that included panmixia, population isolation, change in effective population size, migration, and range expansion. We then identify a smaller subset of models for comparison by generating an expectation of the highest posterior probability that a false model is likely to achieve due to chance and calculate the relative posterior probabilities of only those models that exceed this expected level. A model that parameterized divergence with population expansion and gene flow in one direction, offered the best fit to the P. idahoensis data (in contrast to an isolation only model from the first analysis). Our investigation demonstrates that the determination of which models to include in ABC model choice experiments is a vital component of model-based phylogeographic analysis.
Sampling locality is more detectable than taxonomy or ecology in the gut microbiota of the brood-parasitic Brown-headed Cowbird (Molothrus ater)
Sarah M. Hird, Bryan C. Carstens, Steven W. Cardiff, Donna L. Dittmann, Robb T. Brumfield. 2014. PeerJ 2:e321. DOI: 10.7717/peerj.321
Abstract
Brown-headed Cowbirds (Molothrus ater) are the most widespread avian brood parasite in North America, laying their eggs in the nests of approximately 250 host species that raise the cowbird nestlings as their own. It is currently unknown how these heterospecific hosts influence the cowbird gut microbiota relative to other factors, such as the local environment and genetics. We test a Nature Hypothesis (positing the importance of cowbird genetics) and a Nurture Hypothesis (where the host parents are most influential to cowbird gut microbiota) using the V6 region of 16S rRNA as a microbial fingerprint of the gut from 32 cowbird samples and 16 potential hosts from nine species. We test additional hypotheses regarding the influence of the local environment and age of the birds. We found no evidence for the Nature Hypothesis and little support for the Nurture Hypothesis. Cowbird gut microbiota did not form a clade, but neither did members of the host species. Rather, the physical location, diet and age of the bird, whether cowbird or host, were the most significant categorical variables. Thus, passerine gut microbiota may be most strongly influenced by environmental factors. To put this variation in a broader context, we compared the bird data to a fecal microbiota dataset of 38 mammal species and 22 insect species. Insects were always the most variable; on some axes, we found more variation within cowbirds than across all mammals. Taken together, passerine gut microbiota may be more variable and environmentally determined than other taxonomic groups examined to date.
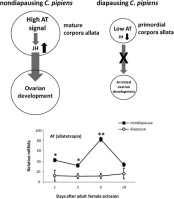
Image: graphical abstract for Kang et al.
Suppression of allatotropin simulates reproductive diapause in the mosquito Culex pipiens
David S. Kang, David L. Denlinger, Cheolho Sim. 2014. Journal of Insect Physiology in press. DOI: 10.1016/j.jinsphys.2014.03.005
Abstract
The cessation of juvenile hormone (JH) production is a key endocrine event that halts ovarian development and hence initiates diapause in females of the mosquito, Culex pipiens. The shutdown in endocrine activity of the corpora allata (CA), the source of JH, was manifested in the smaller size of CA in females reared under short daylengths (diapause) compared to those reared under long daylengths (nondiapause), as well as in low expression of the mRNA encoding allatotropin, the neuropeptide that promotes JH biosynthesis in the CA. Genes encoding both allatotropin and allatostatin were identified in C. pipiens, but only expression levels of allatotropin differed in the two types of females. Knockdown of allatotropin mRNA using RNA interference in females programmed for nondiapause resulted in a cessation of ovarian development akin to diapause. This arrest in development could be reversed with an application of JH. Our results thus suggest that suppression of allatotropin is a critical link in regulating the shutdown of the CA during diapause.
A novel 5-enolpyruvoylshikimate-3-phosphate (EPSP) synthase transgene for glyphosate resistance stimulates growth and fecundity in weedy rice...
Wei Wang, Hui Xia, Xiao Yang, Ting Xu, Hong Jiang Si, Xing Xing Cai, Feng Wang, Jun Su, Allison A. Snow and Bao-Rong Lu. 2014. New Phytologist 202, 679-688. DOI: 10.1111/nph.12428
- Understanding evolutionary interactions among crops and weeds can facilitate effective weed management. For example, gene flow from crops to their wild or weedy relatives can lead to rapid evolution in recipient populations. In rice (Oryza sativa), transgenic herbicide resistance is expected to spread to conspecific weedy rice (Oryza sativa f. spontanea) via hybridization.
- Here, we studied fitness effects of transgenic over-expression of a native 5-enolpyruvoylshikimate-3-phosphate synthase (epsps) gene developed to confer glyphosate resistance in rice. Controlling for genetic background, we examined physiological traits and field performance of crop–weed hybrid lineages that segregated for the presence or absence of this novel epsps transgene.
- Surprisingly, we found that transgenic F2 crop–weed hybrids produced 48–125% more seeds per plant than nontransgenic controls in monoculture- and mixed-planting designs without glyphosate application. Transgenic plants also had greater EPSPS protein levels, tryptophan concentrations, photosynthetic rates, and per cent seed germination compared with nontransgenic controls.
- Our findings suggest that over-expression of a native rice epsps gene can lead to fitness advantages, even without exposure to glyphosate. We hypothesize that over-expressed epsps may be useful to breeders and, if deployed, could result in fitness benefits in weedy relatives following transgene introgression.